How to make mutagenesis primers with Kozane
1. Features
2. How Kozane works
3. The form field
4. The results page
1. Features
Kozane has been developed to make primer design and ordering for site-directed mutagenesis as seamless as possible. It works for methods that rely on primers with the mismatch in the middle of the primer (and not in overhang), such as SAMURAI, Quikchange, PFunkel, and Plasmid-based one-pot saturation mutagenesis.
Scanning options: Predefined alanine, deep mutational, and custom scans.
Cutom mutations: Enter each mutation manually. You can enter amino acids, nucleotides, degenerate codons, insertions, and deletions. You may also introduce several mutations in the same primer.
Quality control: Primers are checked for hairpins, homodimers, GC clamps, and favorable GC concentration.
Matching Tm: The program selects primers with a matching Tm.
Easy ordering: With a single click, all primers selected by the program are copied to your clipboard and you are redirected to the bulk input page of your primer provider.
Surface mutations: Select positions to mutate based on their RSA (relative accessible surface area).

2. How Kozane works
For each mutation, a list of primer candidates is generated. This list contains all possible primers that include a defined set of flanking nucleotides with complementary base pairing on each side of the mutation, but that are not longer than the selected maximum primer length. These primers will subsequently pass through a quality control pipeline. If a primer does not pass a quality control step, it will be removed from the candidate list.
First, primers are discarded from the candidate list if they do not have a Tm within the selected temperature span. The Tm is calculated using Primer3 (version 2.3.7) and subtracting the percentage of the length of the primer that doesn't match the template (indels and degenerate codons are excluded when making this mismatch correction). Next, primers are removed from the list if their Tm with mismatch correction is less than five degrees above its hairpin and homodimer Tm. The hairpin and homodimer Tm is also calculated with Primer3, and while five degrees is the default value, it can be changed under the advance settings. Finally, primers are removed if they don't have a GC clamp; the program checks that the primers end with cytosine or guanine or have two or three cytosines or guanines among the last five 3' bases.
If a Tm has qualified primers for each mutation, these primers will chosen as the final primers. When several Tms have qualified primers for each mutation, the primers for the Tm that has the least extreme maximum GC among its primers will be chosen. If no Tm has qualified primers that covers all mutations, then primers are selected to minimize differences in Tm. You should always end up with primers for all positions; therefore, if no primer passes all the requirements, a handful of the best primers are rescued and a warning will be given on the results page. If no primers pass the Tm requirement, the selected temperature span will be extended for that position.

On the results page, you will be presented with an overview of the results, see potential warnings for the selected primers, and be given the opportunity to study the results closer. By clicking the blue button on the results page, you will automatically copy all selected primers in bulk input format of the primer vendor you have selected and be redirected to their order page where you just have to paste in your primers and place your order.

3. The form field
Specify template and mutations
Template DNA sequence
Enter the sequence that you desire to mutate. The first three bases will correspond to amino acid position 1. The maximum length is 10000 bases.

Mutations
First, select the type of mutations you want to introduce. With Custom mutations, you can essentially make any mutation that you'd like. The Alanine scan will automatically mutate all selected positions to alanine. With the Deep mutational scan, you can make a scan with degenerate codons, with the Custom scan you can essentially make any scan you'd like, and finally, Enter nucleotide positions allows you to specify the mutations you want with nucleotide positions instead of amino acid positions, which is the case for the other options.


Then, in the field below, enter mutations that you want to make. How to use this field depends on the kind type of mutagenesis you selected.
Custom mutations:
Write amino acid positions together with the desired codon or amino acid. Use lowercase letters for nucleotides in complete codon triplets and uppercase letters for amino acids in single letter code. Use * for stop codons. If stating amino acids, the program will select a codon that implies mutating as few bases as possible. Example: 1A 15atg will give you one primer that mutates amino position 1 to alanine and a second primer that mutates amino position 15 to methionine.You can mutate several positions in a primer by writing them in a row. Example: 15AatgC will give you a primer that mutates amino acid position 15 to alanine, 16 to methionine, and 17 to cysteine.
To make insertions, write what you want to insert between two ^. It will be inserted after the amino acid position that is stated. Example: 15^AatgC^ will make a primer that inserts an alanine, a methionine, and a cysteine between amino acid positions 15 and 16.
For deletions, state amino acid positions and use underscores for deletions. Example: 15_ will give you a primer that deletes the amino acid at position 15.
You can make mutations before and after indels. Example: 15atg^G^S will make a primer that changes amino acid positions 15 to a methionine, inserts a glycine between position 15 and 16, and changes positions 16 to a serine. It is not possible to make two different insertions or make both an insertion and a deletion in the same primer.
Alanine scan:
Write the amino acid positions you want to change in the mutations field. A dash can be used for a span of positions. So for example writing 12 15 75-90 will give you primers to mutate amino acid positions 12, 50 and 75 to 90 to alanines. Leaving the field blank will mutate all positions to alanine. It is also possible to state the positions that you don't want to mutate; -(12 15 75-90) will mutate all positions in the template except 12, 15 and 75 to 90. The program will use the codon that requires the fewest mutations. Use 'custom scan' if you want a specific codon.Deep mutational scan:
First, write a degenerate codon followed by the desired positions. So nnk 12 15 75-90 will mutate amino acid positions 12, 50 and 75 to 90 to nnk. Exclude stating positions if you want to mutate all positions. It is also possible to state the positions that you don't want to mutate; nnk -(12 15 75-90) will mutate all positions in the template except 12, 15 and 75 to 90. The hairpin/homodimer QC will be turned off with degenerate codons and such codons will be excluded in the Tm calculations.Custom scan:
Write an amino acid or codon followed by the amino acid positions that you want to mutate. Example: C 12 15 75-77 will mutate position 12, 15, 75, 76 and 77 to cysteine and gca -(12 15 75-77) will mutate all positions except 12, 15 75, 76 and 77 to alanine with a gca codon. This can, for example, be used to do an alanine scan with a specific codon.Enter nucleotide positions:
With this option, you state the nucleotide positions you want to mutate. Example: 1a 15gc will change the first nucleotide position to adenine in one primer, and another primer will mutate nucleotide positions 15 to guanine and 16 to a cytosine.You can also make insertions and deletions with this option, although it is more straightforward with "Custom mutations". Insertions and deletions must be made so that you'll end up with a complete codon triplet, i.e., you cannot make an insertion with only two nucleotides or delete only one nucleotide of a codon. It is not possible to make two different insertions or make both an insertion and a deletion in the same primer. For insertions, the insertion will happen after the codon that the defined nucleotide position belongs to. Example: 20c^ctg^ will mutate nucleotide position 20 to c and insert a proline between amino acid positions 7 and 8.
Note that position 20 is the second base of the codon. If you would write 20cg^ctg^ you would mutate position 20 to c, 21 to g, and insert a proline between amino acid positions 7 and 8. Writing 20cgg^ctg^ implies that the last base before the insertion is part of amino acid position 8. In this case, the insertion will still be made between amino acid positions 7 and 8 unless all three bases of the latter codon are included 20cggtc^ctg^. In that case, the insertion will end up after amino position 8. This might be a bit confusing and is why working in frame with "Custom mutations" is more straightforward.
Regarding deletions, for "Enter nucleotide positions" three underscores are used to delete a codon. Example: 20cg___tg will give you a primer that mutates nucleotide position 20 to c, 21 to g, it will delete the following codon and mutate position 25 to t and 26 to g.
Surface mutations:
Use this option to select which positions to mutate based on surface exposure. This is done based on a PDB ID and the xssp web server API, using the DSSP output. ACC values from the DSSP result are subsequently recalculated into RSA values with the theoretical reference values from Tien et al (2013). Cite both xssp and Tiam accordingly if you use this option.You select a PDB ID, enter a nucleotide sequence and state which amino acid chains this sequence corresponds to in the PDB entry. An RSA threshold (%) is set and positions with an RSA above this value are determined and selected for mutagenesis. You also have the option to exlude certain amino acids for mutagensis even though their RSA values are above the threshold. Note that some residues may be missing in the amino acid sequence for the PDB entry; these will automatically be inserted based on your nucleotide sequence. Verify in the detailed results in the flash message that shows up after the calculations are done to assure that the PDB sequence matches your nucleotide sequence. Inserted amino acids will automatically be selected for mutagenesis unless they are among the amino acids to be excluded. Once the calculations are done, the "Enter mutations and their amino acid positions" will have been prepared with the positions that should be mutated according to your input. The field will begin with an A, i.e. it will give you a standard alanine scan, but you may change this "A" to anything you want á la the "Custom scan" option.
Before you generate your primers...

Before you begin generating your primers, you have the option to preview the mutations you have entered using the "preview template with mutations" button that is situated right below the mutations field. This preview will show you the DNA template sequence you entered, its translated sequence, the amino acid positions, and all the mutatons that will be introduce.
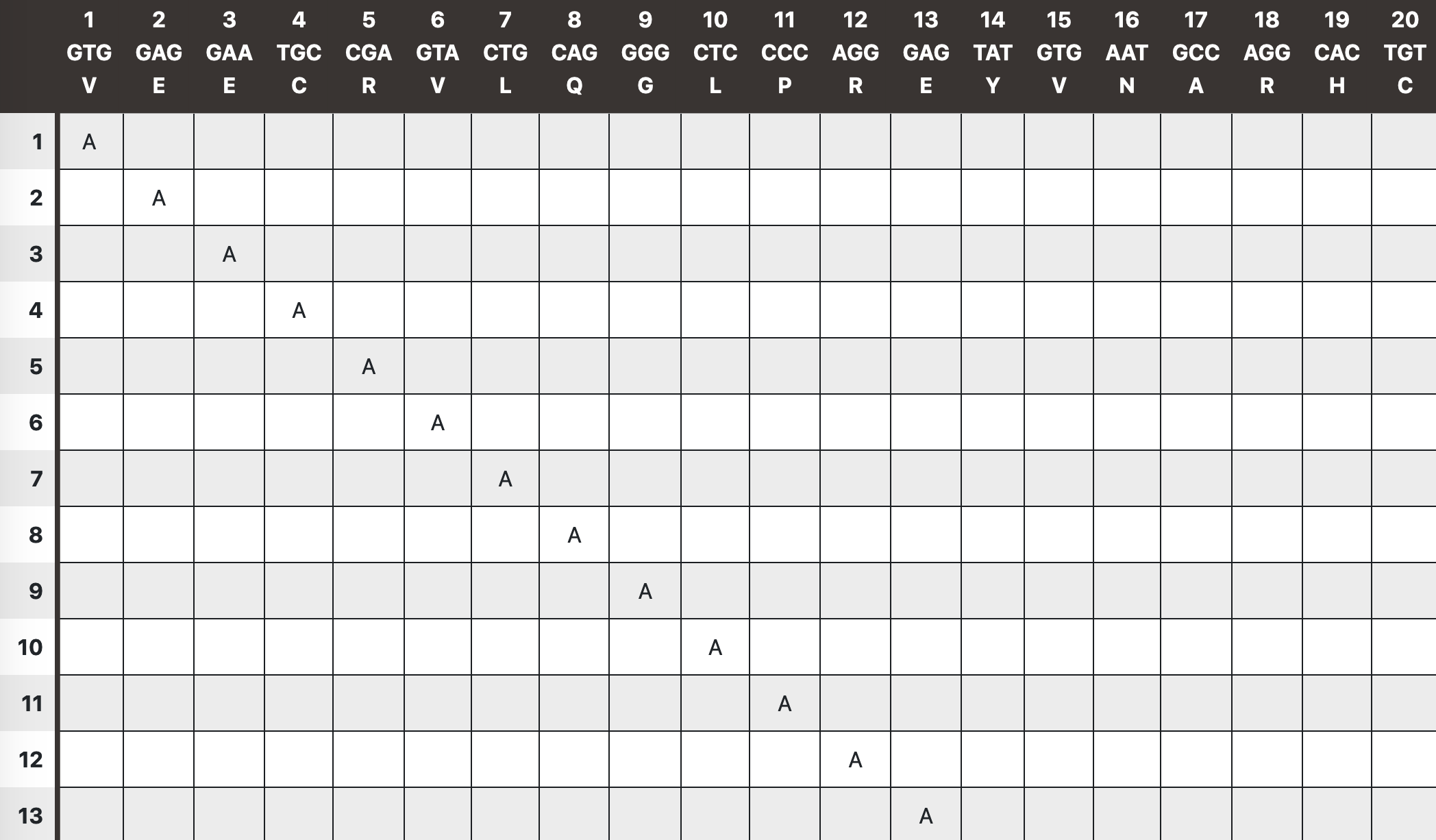
Upstream sequence & Downstream sequence
If mutations are to be introduced close to the ends of the template sequence, a few extra bases must be provided to allow the primers to also hybridize outside of the template sequence. Fifty bases should typically be enough (depends on the "primer length" and how many "flanking" bases you want to have). You will be asked to add more bases if needed.
Set primer properties
Min and Max Tm
The program will search for primers between the temperatures set here. A recommendation is to start with a wide temperature span and then narrow it down if needed. If no primer would be found within the temperature span for a specific mutation, the program will widen the span for that position until a qualified primer is found. Min and Max cannot be the same number.
Max length primer
The maximum number of bases a primer is allowed to have.
Min flanking positions
The minimum number of bases a primer must have on both sides of the mismatch.
Primer direction
Choose forward, reverse or both.
The buttons
Return primer properties to defaults
Reverts changes you have made to the primer properties to the defaults.
Use Agilent Quikchange preset
If you press this button, the primer properties are set to mimic the recommendations in the Quikchange protocol. More specifically, min Tm is set to 78 degrees (max Tm to 85 although no exact value is mentioned in the protocol), max length primer is set to 45, min flanking positions to 10, and primer direction to "make both" forward and reverse primers. Under the advanced settings, Tm method is set to "Agilent Quikchange", which implies the following calculations:
Tm = 81.5 + 0.41 * %GC - 675/(primer length)-%mismatch
Or the one below if using insertion, deletions or degenerate codons are involved.
Tm = 81.5 + 0.41 * %GC - 675/(primer length excluding indels or degenerate codons)-%mismatch
A slight difference compared to the Agilent protocol is that %GC and %mismatch are used with decimal values and not whole numbers. Furthermore, the hairpin/homodimer Tm control is turned off. Feel free to turn it on in the advanced settings; it can be set quite high as the just mentioned Tm calculation gives relatively high values compared to "santalucia" and "breslauer". The rest of the options under advance settings can be ignored unless the hairpin/homodimer QC is turned on.
Show advance options
The advanced settings let you control how many degrees the primer Tm (with mismatch calculation) must be above its hairpin and homodimer Tm. The advanced settings also allow you to control the monovalent cation concentration, divalent cation concentration, dNTP concentration, DNA concentration, Tm calculation method, salt correction method, the maximum length for nearest-neighbor calcs, the maximum size of loops in the structure and simulation temperature for dG calcs. Consult the Primer3 documentation for more information about these options.
Choose ordering alternatives
Primer provider
State from where you will buy your primers. This will prepare your "bulk input" file and provide you with a direct link on the "Results page" to your vendor.
Primer name
Primers will be given a name based on what you write here plus the position, Tm and direction of the primer. If you write "myprimer" in this field, a primer might look something like myprimer_Y50A_tm58R. Y50A means that the primer will change a tyrosine to an alanine at amino acid position 50, tm58 tells you it has a Tm of 58 degrees, and R stands for reverse direction (would be F for forward a forward primer).
5' ext/overhang
Whatever you write here will be added at the beginning of your primer sequences. You can, for example, write /5Phos/ if you order from IDT and want a 5'Phosphorylation on your primers.
4. The results page
Results overview and warning messages
On the results page, you will be shown which Tm (or Tms) that was picked for your selected primers. You will also be informed if one or more of the selected primers did not fulfill the Tm, hairpin, homodimer or GC clamp criteria. In the end, among all primers that passed all QC requirements, the program will for each mutation select the primer that is the closest to GC concentration 50 %. If it is a particularly GC-rich region, you might get an error that the primer GC-content is above 65 %. A warning will also be raised if a mutations does not result in a change in amino acid.
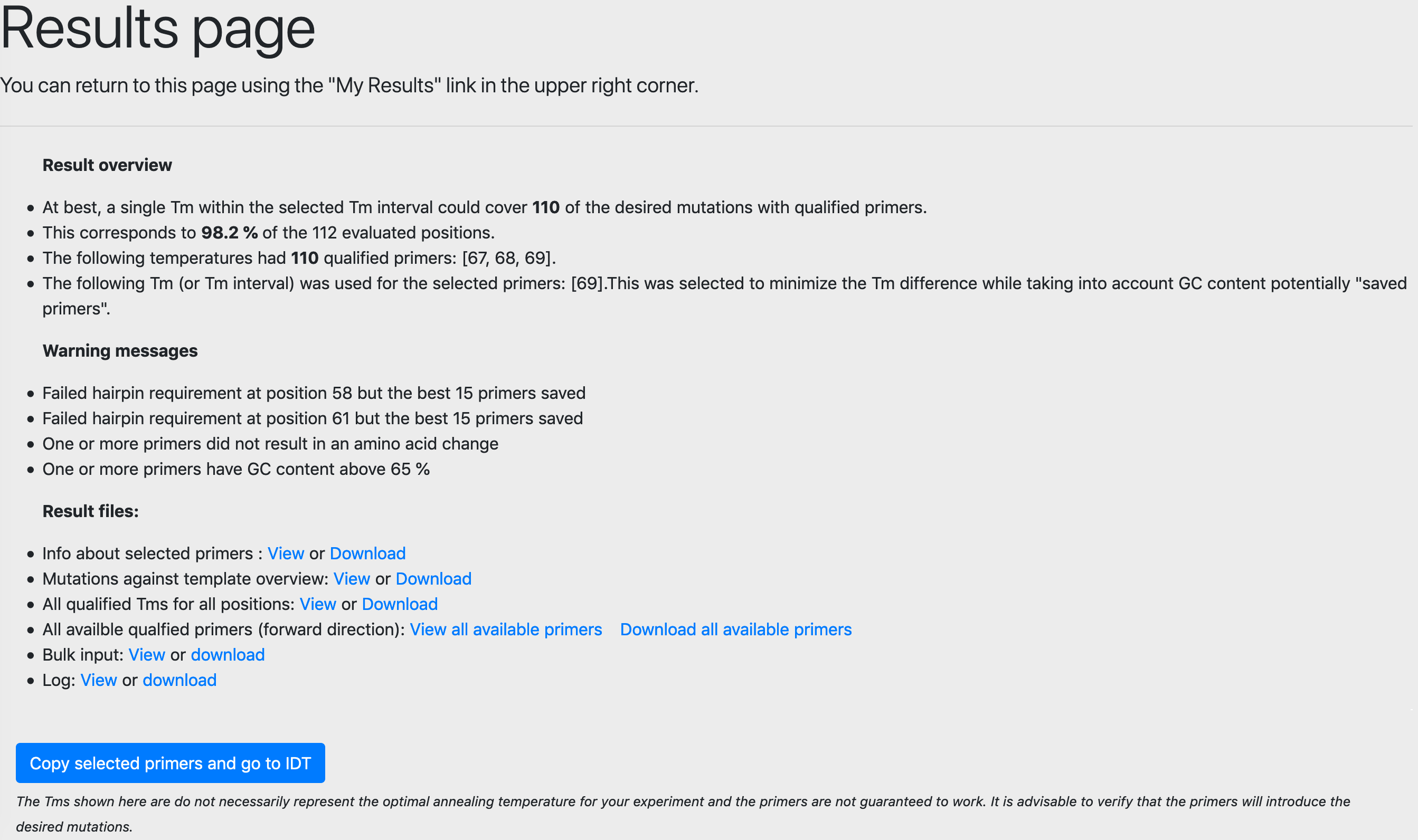
Result files
Info about selected primers
The primer list shows a detailed description of the primers that were selected.
| AA position | new codon | original aa | new aa | name | final primer | with extension | against template | length | tm with mismatch calculation | hairpin tm | homodimer tm | tm without mismatch correction | percent GC | reverse complement name | reverse complement primer |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GCG | V | A | primer_V1A_tm70R | ATTCCTCCGCATTCACATACTCCCTGGGGAGCCCCTGCAGTACT | ATTCCTCCGCATTCACATACTCCCTGGGGAGCCCCTGCAGTACT | AGTACTGCAGGGGCTCCCCAGGGAGTATGTGAATGTGGAGGAAT | 44 | 69.6 | 55.4 | 25.7 | 71.9 | 56.8 | primer_V1A_tm70F | AGTACTGCAGGGGCTCCCCAGGGAGTATGTGAATGCGGAGGAAT |
| 2 | GCG | E | A | primer_E2A_tm69R | TGCAGTACTCGGCATTCCGCCACATTCACATACTCCCTGGGGAG | TGCAGTACTCGGCATTCCGCCACATTCACATACTCCCTGGGGAG | CTCCCCAGGGAGTATGTGAATGTGGAGGAATGCCGAGTACTGCA | 44 | 69.3 | 60.4 | 24.5 | 71.6 | 56.8 | primer_E2A_tm69F | CTCCCCAGGGAGTATGTGAATGTGGCGGAATGCCGAGTACTGCA |
| 3 | GCA | E | A | primer_E3A_tm69R | TGCAGTACTCGGCATGCCTCCACATTCACATACTCCCTGGGGAG | TGCAGTACTCGGCATGCCTCCACATTCACATACTCCCTGGGGAG | CTCCCCAGGGAGTATGTGAATGTGGAGGAATGCCGAGTACTGCA | 44 | 69.1 | 45.5 | 29.4 | 71.4 | 56.8 | primer_E3A_tm69F | CTCCCCAGGGAGTATGTGAATGTGGAGGCATGCCGAGTACTGCA |
| 4 | GCC | C | A | primer_C4A_tm69R | ATACTCCCTGGGGAGCCCCTGCAGTACTCGGGCTTCCTCCACAT | ATACTCCCTGGGGAGCCCCTGCAGTACTCGGGCTTCCTCCACAT | ATGTGGAGGAATGCCGAGTACTGCAGGGGCTCCCCAGGGAGTAT | 44 | 69.1 | 57.5 | 38.7 | 73.7 | 61.4 | primer_C4A_tm69F | ATGTGGAGGAAGCCCGAGTACTGCAGGGGCTCCCCAGGGAGTAT |
| 5 | GCA | R | A | primer_R5A_tm69R | CCCTGGGGAGCCCCTGCAGTACTGCGCATTCCTCCACATTCACAT | CCCTGGGGAGCCCCTGCAGTACTGCGCATTCCTCCACATTCACAT | ATGTGAATGTGGAGGAATGCCGAGTACTGCAGGGGCTCCCCAGGG | 45 | 69.3 | 50.2 | 35.0 | 73.7 | 60.0 | primer_R5A_tm69F | ATGTGAATGTGGAGGAATGCGCAGTACTGCAGGGGCTCCCCAGGG |
| 6 | GCA | V | A | primer_V6A_tm69R | AGCCCCTGCAGTGCTCGGCATTCCTCCACATTCACATACTCC | AGCCCCTGCAGTGCTCGGCATTCCTCCACATTCACATACTCC | GGAGTATGTGAATGTGGAGGAATGCCGAGTACTGCAGGGGCT | 42 | 69.0 | 38.8 | 25.9 | 71.4 | 57.1 | primer_V6A_tm69F | GGAGTATGTGAATGTGGAGGAATGCCGAGCACTGCAGGGGCT |
| 7 | GCG | L | A | primer_L7A_tm70R | TGGCATTCACATACTCCCTGGGGAGCCCCTGCGCTACTCGGCATT | TGGCATTCACATACTCCCTGGGGAGCCCCTGCGCTACTCGGCATT | AATGCCGAGTACTGCAGGGGCTCCCCAGGGAGTATGTGAATGCCA | 45 | 69.9 | 55.4 | 25.7 | 74.3 | 60.0 | primer_L7A_tm70F | AATGCCGAGTAGCGCAGGGGCTCCCCAGGGAGTATGTGAATGCCA |
| 8 | GCG | Q | A | primer_Q8A_tm70R | TGGCATTCACATACTCCCTGGGGAGCCCCGCCAGTACTCGGCATT | TGGCATTCACATACTCCCTGGGGAGCCCCGCCAGTACTCGGCATT | AATGCCGAGTACTGCAGGGGCTCCCCAGGGAGTATGTGAATGCCA | 45 | 69.8 | 55.7 | 25.7 | 74.2 | 60.0 | primer_Q8A_tm70F | AATGCCGAGTACTGGCGGGGCTCCCCAGGGAGTATGTGAATGCCA |
| 9 | GCG | G | A | primer_G9A_tm70R | TGGGGAGCGCCTGCAGTACTCGGCATTCCTCCACATTCACATAC | TGGGGAGCGCCTGCAGTACTCGGCATTCCTCCACATTCACATAC | GTATGTGAATGTGGAGGAATGCCGAGTACTGCAGGGGCTCCCCA | 44 | 69.6 | 37.9 | 22.3 | 71.8 | 56.8 | primer_G9A_tm70F | GTATGTGAATGTGGAGGAATGCCGAGTACTGCAGGCGCTCCCCA |
| 10 | GCC | L | A | primer_L10A_tm70R | TGGCATTCACATACTCCCTGGGGGCCCCCTGCAGTACTCGGCATT | TGGCATTCACATACTCCCTGGGGGCCCCCTGCAGTACTCGGCATT | AATGCCGAGTACTGCAGGGGCTCCCCAGGGAGTATGTGAATGCCA | 45 | 69.8 | 52.0 | 37.4 | 74.3 | 60.0 | primer_L10A_tm70F | AATGCCGAGTACTGCAGGGGGCCCCCAGGGAGTATGTGAATGCCA |
Scroll to see the full table
Codon: Indicates the amino acid position. The first three bases of your template sequence will mark codon 1.
Mutation: The nucleotides after mutation.
Original aa: The original amino acids.
New aa: The amino acids after mutation
Name: The given primer name.
Final primer: The selected primer.
With extension: The selected primer including potential 5' extension or overhang.
Against template: What the template looks like where the primer binds, also highlighting mutated bases.
Length: The primer length.
Tm with mismatch calculation: The estimated Tm considering mismatches. This is calculated by taking the primer Tm (using Primer3) and subtracting the percentage of the length of the primer that doesn't match the template. Degenerate positions are not included in the calculation.
Hairpin Tm: The hairpin Tm according to Primer3.
Homodimer Tm: The homodimer Tm according to Primer3.
Tm without mismatch correction: The primer Tm according to Primer3. Degenerate positions are not included in the calculation
Percent GC: The primer GC content in percent.
Reverse complement name: The name for the reverse complement of the selceted primer.
Reverse complement primer: The reverse complement of the selected primer.
All qualified Tms for all positions
This table shows the Tms that had qualified primers for each codon and summarizes how many qualified primers each Tm had.
Mutations against template overview
Same as "preview template with mutations" button, i.e., it will show you the DNA template sequence you entered, its translated sequence, the amino acid positions, and all the mutatons that will be introduce.
All availble qualfied primers
This table shows the primer sequences for the best primer for each Tm and mutations. Note that this table also will include primers that was "rescued". So if no primer for a position passed the hairpin check, the best 15 primers considering the hairpin Tm will be "rescued". Such rescued primers will show up in this table but not in the primer overview table.
Bulk input
Each primer vendor has a method to upload multiple primers by copy and pasteing text or by uploading a file. This file is prepared with the formatting used by the primer vendor you picked. Note that the content on bulk input page will automatically be copied to your clipboard upon pressing the blue button on the results page. The button will also take you to the ordering page where you can select bulk input and paste (Ctrl-V/cmd-V) your primers.

Log file
The log files contains your input parameters. This makes the log file especially important if you encounter a bug. If so, please report your import parameters to facilitate finding and exterminating the bug. The log file also shows how many primers survive each of the quality control checks for each codon.